common file formats in bioinformatics
最初,简单的文本文件(比如Txt 文件)用于存储序列数据使用单核苷酸或氨基酸代码。
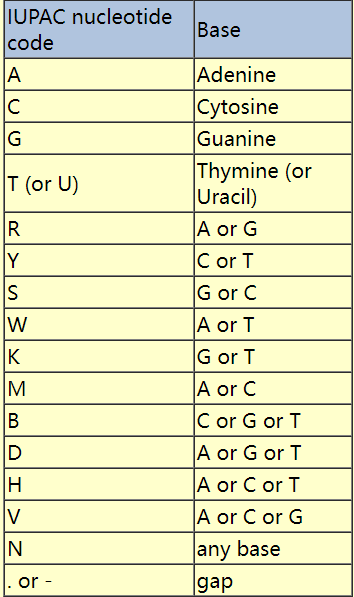
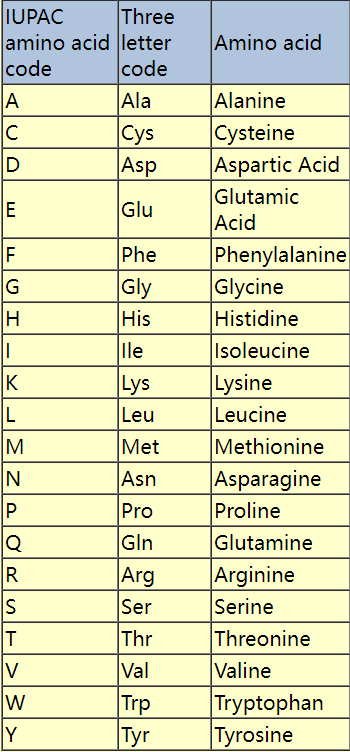
就比如
ATCGNNNRYSW但是纯文本信息不能对序列的染色体、质量、功能等信息进行注释,所以需要开发一些对应的格式
- The Different Bioinformatics File Types
- Why are There so Many Different Types?
- File Formats and BLAST
- Conclusion
- File Format FAQs
- References
The Different Bioinformatics File Types
FASTA

- fasta格式,生物信息学中的标准文件格式,发明于1988年,用来进行 DNA 或蛋白质序列的敏感序列比对
- fasta格式,使用核苷酸或氨基酸单字母来表示DNA或蛋白质序列
- fasta格式,第一行是序列标识符,前面有一个“>”,接上序列信息,公共数据库里有标准的格式。第二行就是核苷酸或氨基酸的单字母来表示序列,由于早期DECVCT的终端,一行字符的数量有限,所以每行Fasta字符数一般限制在80字符以内。
- nucleic acids (.fna)核酸
- nucleotide coding regions (.ffn)核酸编码区
- amino acids (.faa)氨基酸
- non-coding RNAs (.frn)非编码RNA
- fasta文件可以包含一个或多个序列,ClustalW等工具可以使用带有多个序列的FASTA文件来生成对齐
FASTQ

- FASTQ格式是一种保存生物序列(通常为核酸序列)及其测序质量得分信息的文本格式。序列与质量得分皆由单个ASCII字符表示。该格式最初由维尔康姆基金会桑格研究所开发,旨在将FASTA格式序列及其质量数据集成在一起。而目前,FASTQ格式已经成为了保存高通量测序结果的事实标准。
- next-generation sequencing二代测序的发明,开发了FASTQ格式
- 第一行以@开头,之后为序列的标识符以及描述信息(与FASTA格式的描述行类似),标本格式和Illumina测序仪相关,包括唯一的仪器名称、流池通道等。
- 第二行为序列信息,和FASTA一样
- 第三行以+开头,之后可以再次加上序列的标识及描述信息(可选)
- 第四行为质量得分信息,与第二行的序列相对应,长度必须与第二行相同
- .fastq, .sanfastq, or .fq都是FASTQ
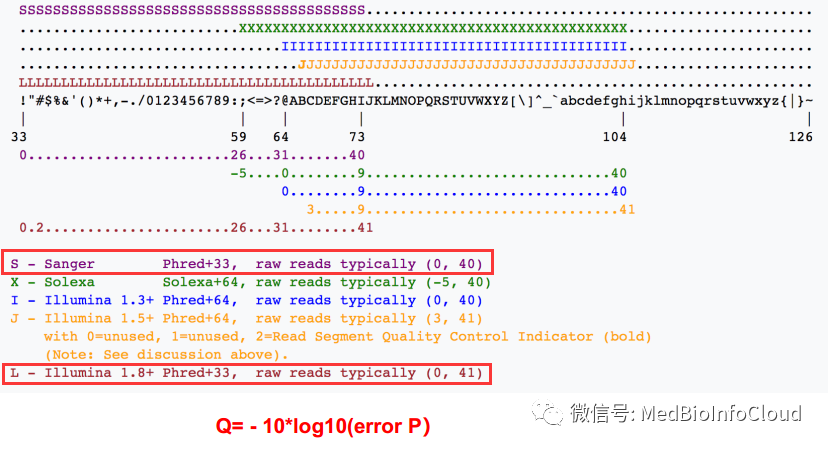
从左到右代表从低到高的质量得分
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~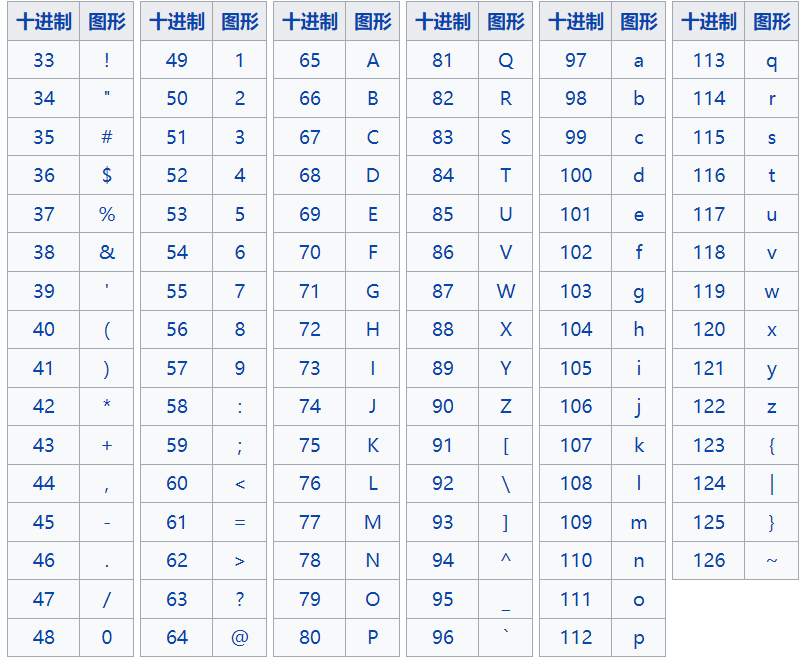
所以,FASTQ的序列信息一般就是一行结束,最初的时候桑格那边用过多行,但是信息提取不方便
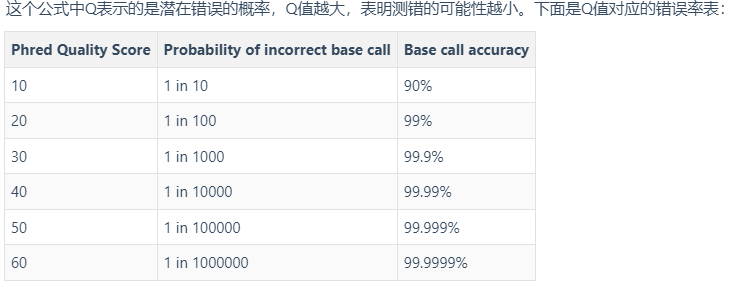
Q= -log10p
Q= 20,p=0.01,碱基的精确度就是99%
假设某一核苷酸对应的第四行质量信息是“5”,按照上表换成10进制就是53,如果采用的是Sanger测序,那采用的就是Phread+33,Q = 53 - 33 = 20
除了Phread+33,还有Solexa+64等等
Alignment formats对齐格式
BAM
生信数据文件常见格式介绍-2 · XTAO Achelous
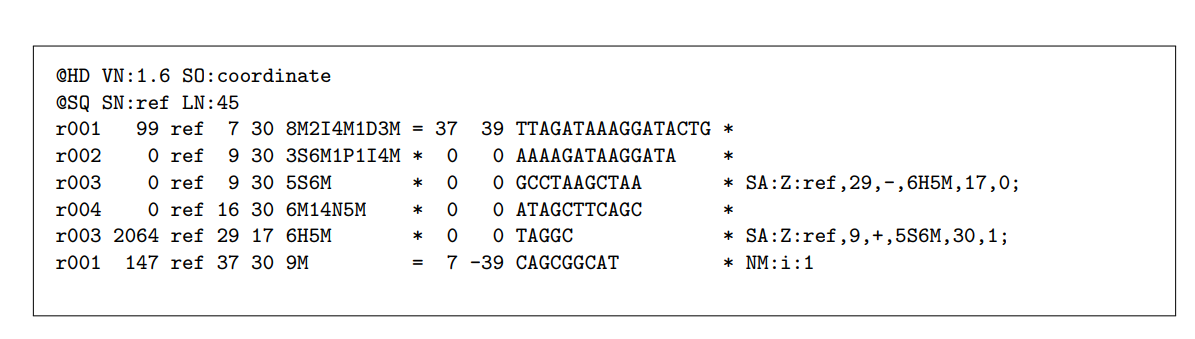
BAM文件(扩展名为. BAM)与SAM文件密切相关,SAM文件是用制表符分隔的文本文件,用于存储序列对齐数据。BAM文件格式优于SAM文件格式的优点是,它是一个压缩的二进制版本,大小更小,可以索引,使它们成为序列比对信息存储的理想选择,并首选于整合基因组学查看器。
- BAM包含一个头和一个主体,标头存储有关序列的信息,前面有一个“@”符号。主体包含关于每个序列如何与特定的参考序列对齐的信息
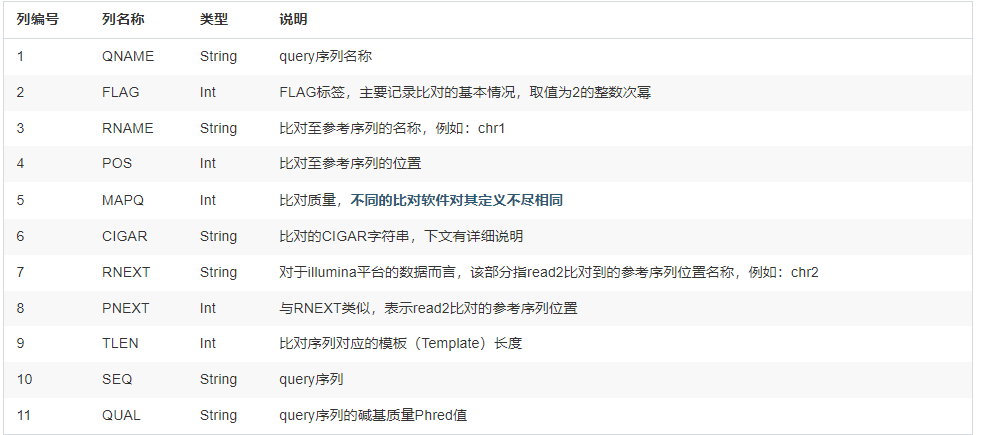
- 每个对齐行包括11个数据字段,包括Phred评分、描述对齐的字符串(称为CIGAR)和其他元数据。
SAM
SAM文件代表序列比对/MAP,并以. SAM文件扩展名表示,最初源自一款名为SAMtools的生物信息学软件,这是一款用于查看比对的开源程序。
- sam 文件头部信息以
@作为开头,对比对结果的meta信息进行记录,主要涵盖了文件标准格式版本(VN)、比对中使用的参考序列信息(SQ)、测序数据分组信息(RG)、比对或后期处理使用的程序信息(PG)等。 sam文件的比对信息中,每一行是由固定的11列的组成的。其信息内容见下表:每行可以追加可选信息。该部分信息以TAG:TYPE:VALUE形式存储
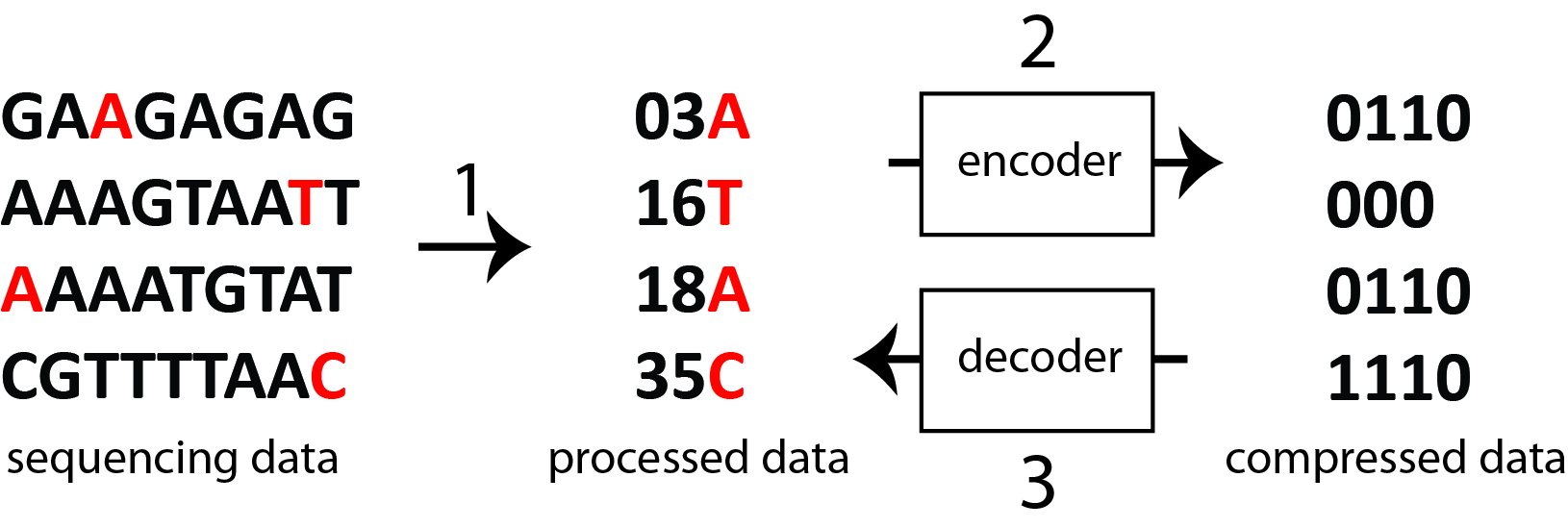
从SAM压缩到BAM
CRAM
CRAM文件是另一种与BAM文件格式相关的文件类型,它是BAM文件的重新构造版本,支持无损压缩
SRA⭐
SRA(Sequence ReadArchive)数据库是用于存储二代测序的原始数据,包括 454,Illumina,SOLiD,IonTorrent,Helicos 和 CompleteGenomics。除了原始序列数据外,SRA现在也存在raw reads在参考基因的比对信息。
根据SRA数据产生的特点,将SRA数据分为四类:
- Studies– 研究课题
- Experiments– 实验设计
- Runs– 测序结果集
- Samples– 样品信息
SRA中数据结构的层次关系为:Studies->Experiments->Samples->Runs.
- Studies是就实验目标而言的,一个study 可能包含多个Experiment。
- Experiments包含了Sample、DNA source、测序平台、数据处理等信息。
- 一个Experiment可能包含一个或多个runs。
- Runs 表示测序仪运行所产生的reads。
SRA数据库用不同的前缀加以区分:
- ERP或SRP表示Studies;
- SRS 表示 Samples;
- SRX 表示 Experiments;
- SRR 表示 Runs;
Stockholm formats斯德哥尔摩格式
VCF
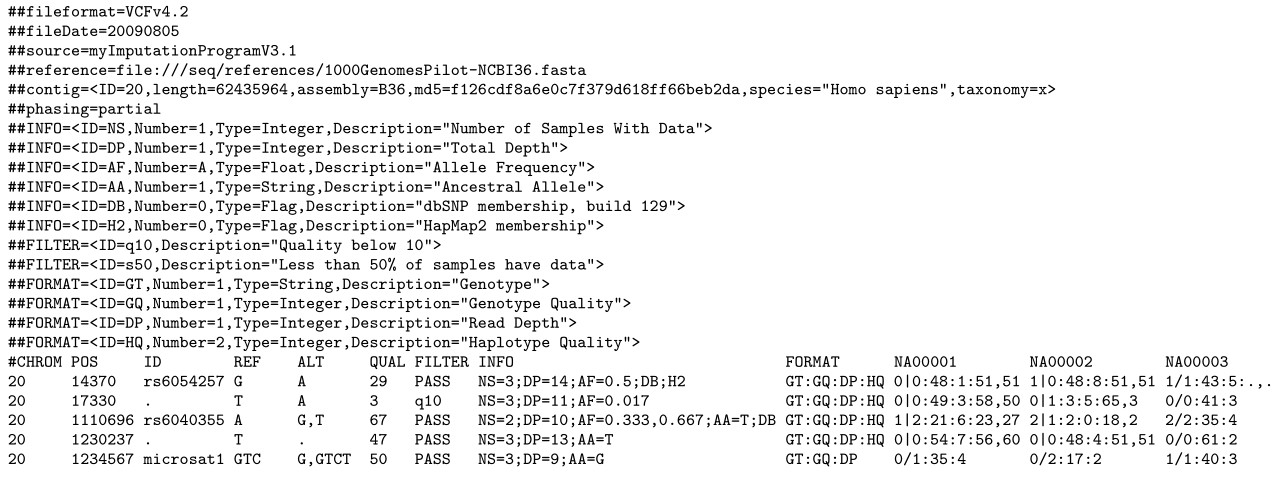
VCF (Variant Calling Format; file extension .vcf) files可变呼叫格式;文件扩展名为.vcf)文件存储基因序列变异,如单核苷酸多态性(SNPs),并用于基因分型项目。它包含一个以“##”字符串开头的元数据头。VCF文件的最佳实践建议在头文件中描述正文中使用的INFO、FILTER和FORMAT条目。
标题后面是主体,由8个必列组成,每个标识符对应一个列
Generic feature formats通用特性格式
GFF
A GFF (general feature format; file extension .gff2 or .gff3) 一种GFF(一般特征格式;文件扩展名。gff2或。gff3)描述了组成基因的各种序列元素,是注释基因组的标准方法它定义了GFF文件主体中基因的特征,包括转录本、调控区域、未翻译区域、外显子、内含子和编码序列。与VCF一样,它使用带有“##”字符串的头区域来包含元数据。
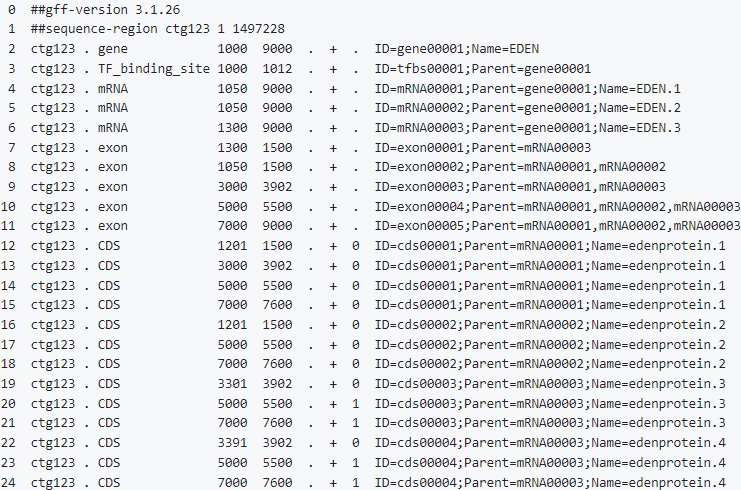
GTF
GTF(基因转移格式)文件类型与GFF文件共享相同的格式,尽管它专门用于定义基因和转录相关的特征上面描述的属性/组字段在GTF中用于包含gene_id或transcript_id值,这两个值分别是基因组源或预测转录本的唯一标识符。
Unlabeled formats
BED(浏览器可扩展数据)文件格式包括可以在基因组浏览器中可视化的序列信息;一个被称为注释轨迹的特性BED文件以制表符分隔,包括12个字段(列)的数据。要正确读取每个文件的行,列必须是一致的。
PDB文件格式包含原子坐标,蛋白质数据库用于存储3D蛋白质结构。有关格式的更多信息,请参阅pyMOL(参见完整指南)
PED。Ped文件扩展名)是一种用于谱系分析的文件格式,它在不同的样本之间创建了家族关系它与PLINK命令行生物信息学程序一起使用。
MAP。map文件扩展名)是使用PLINK程序时伴随PED文件格式的一种文件格式它包含不同的信息。
Why Are There So Many Different Types?
生成和使用测序数据的许多不同方法已经产生了上面描述的序列文件格式,这些方法或者说软件,产生了这些数据,为了和软件兼容,增加可读性、便于处理,便于储存
FASTA和FASTQ用于存储原始测序数据,但FASTQ文件也保存质量数据。FASTA还可以存储DNA、RNA和蛋白质序列,而FASTQ通常只包含DNA序列。
SAM、BAM和CRAM文件的相似之处在于它们存储序列对齐数据,但压缩状态不同
BED、VCF和GFF/GTF不存储原始序列数据,而是有各种DNA特征注释。这些特性注释因文件类型而异。
通过介绍生物信息学中的标准文件格式,您应该了解每种文件类型的结构、生物数据和用途。掌握如何在实际的、真实的环境中使用这些文件的最好方法是开始定期使用它们,并熟悉大量可用的生物信息学工具和软件。
flat format什么是生物信息学中的平面格式? 平面文件格式是每行只有一条记录的表格。FASTA和其他文件格式是生物信息学中平面文件格式的一个例子。
data types 生物信息学中的数据类型可以是DNA序列、RNA序列、氨基酸序列、甲基化序列、三维蛋白质结构等等。
这些不同的文件类型可用于与其他生物信息学软件兼容或提高存储效率。

