参考链接
国家微生物科学数据中心-CheckM使用说明内有视频及PDF讲解,还可线上测试。
参考微信公众号宏基因组文章
Background
CheckM首先基于完整的已测序细菌基因组作为参考基因组,构建基因组的进化树,构建每个谱系(可以理解为一类物种)的单拷贝基因集(管家基因)(single copy genes,SCGs,为什么是单拷贝?因为这样可以开展基因组混合程度、污染程度等的评估)。在使用时,将我们的Bin与参考基因组一起建树,基于进化关系找到Bin的参考物种,然后结合参考物种的单拷贝基因集,计算两个重要指标
Completeness,完整度,Bin基因与对应SCGs相比,基因数量是否完整,取值[0,100%],数值越大,表示Bin质量越好; Contamination,污染度,Bin基因包含多个物种的SCGs,即一个Bin存在多个物种的程度,取值[0,100%],数值越小,表示Bin质量越好。
获得每个bin的污染度、完整度信息后,挑选高质量的bin进行物种、功能注释。再后续分析中,并没有固定标准。需要的数量多,则放宽阈值;需要的数量少,则提升阈值。其中,最常用的指标是污染度小于10%,且完整度大于80%。大家可以在这个基础上上下调整。
得到了组装的bin的数据,CheckM下载完成,开始分析
CheckM的工作流程
lineage-specific(世系特异性)==【推荐方法】==
checkm lineage_wf <bin folder> <output folder>
##根据基因组在参考基因组发育树中的位置,来推断它的single-copy基因集,需要有checkM的数据库taxonomic-specific(物种分类特异性)
checkm taxonomy_wf <rank> <taxon> <bin folder> <output folder> <rank>: phylum; <taxon> : Cyanobacteria
##自己知道自己的数据来自哪个门,什么科的- custom marker genes(自行指定基因maker)
checkm analyze <custom HMM file> <bin folder> <output folder>
checkm qa <custom HMM file> <output folder>
##自己预测了基因maker,<custom HMM file>就是预测的结果【推荐方法】
lineage-specific(世系特异性)(使用该方法需下载checkM的参考数据集,并设置安装在相应位置)
##下载数据库,并解压设置路径
wget -c https://data.ace.uq.edu.au/public/CheckM_databases/checkm_data_2015_01_16.tar.gz
tar -zxvf checkm_data_2015_01_16.tar.gz
checkm data setRoot $PATH/checkm_data==主要操作命令==
checkm lineage_wf -h的详细参数(主要用这个)
$ checkm lineage_wf -h
usage: checkm lineage_wf [-h] [-r] [--ali] [--nt] [-g] [-u UNIQUE] [-m MULTI]
[--force_domain] [--no_refinement]
[--individual_markers] [--skip_adj_correction]
[--skip_pseudogene_correction]
[--aai_strain AAI_STRAIN] [-a ALIGNMENT_FILE]
[--ignore_thresholds] [-e E_VALUE] [-l LENGTH]
[-f FILE] [--tab_table] [-x EXTENSION] [-t THREADS]
[--pplacer_threads PPLACER_THREADS] [-q]
[--tmpdir TMPDIR]
bin_dir output_dir
Runs tree, lineage_set, analyze, qa #跑tree、lineage_set、analyze、qa这四个操作
positional arguments:##位置参数
bin_dir directory containing bins (fasta format)###你组装得到bin的目录
output_dir directory to write output files##写入输出文件的目录
optional arguments:##可选参数
-h, --help show this help message and exit##就是我不了解参数,救命
-r, --reduced_tree use reduced tree (requires <16GB of memory) for determining lineage of each bin##使用精简树(需要小于16 GB的内存)来确定每个箱的谱系
--ali generate HMMER alignment file for each bin##为每个条柱生成HMMER对齐文件
--nt generate nucleotide gene sequences for each bin##为每个bin生成核苷酸基因序列
-g, --genes bins contain genes as amino acids instead of nucleotide contigs##bin中含有氨基酸而不是核苷酸重叠群的基因
-u, --unique UNIQUE minimum number of unique phylogenetic markers required to use lineage-specific marker set (default: 10)##使用谱系特定标记集所需的唯一系统发育标记的最小数量(默认值:10)
-m, --multi MULTI maximum number of multi-copy phylogenetic markers before defaulting to domain-level marker set (default: 10)##默认设置为域级标记集之前的多拷贝系统发育标记的最大数量(默认值:10)
--force_domain use domain-level sets for all bins##对所有bin使用域级别集
--no_refinement do not perform lineage-specific marker set refinement##不执行特定于谱系的标记集细化
--individual_markers treat marker as independent (i.e., ignore co-located set structure)##将标记视为独立的(即忽略共存的集合结构)
--skip_adj_correction
do not exclude adjacent marker genes when estimating contamination##在估计污染时不排除相邻的标记基因
--skip_pseudogene_correction
skip identification and filtering of pseudogenes##假基因的跳跃识别与过滤
--aai_strain AAI_STRAIN
AAI threshold used to identify strain heterogeneity (default: 0.9)##用于识别菌株异质性的AAI阈值(默认值:0.9)
-a, --alignment_file ALIGNMENT_FILE
produce file showing alignment of multi-copy genes and their AAI identity##生成显示多拷贝基因的比对及其AAI身份的文件
--ignore_thresholds ignore model-specific score thresholds##忽略特定于型号的分数阈值
-e, --e_value E_VALUE
e-value cut off (default: 1e-10)##E值截止(默认:1e-10)
-l, --length LENGTH percent overlap between target and query (default: 0.7)##目标和查询之间的重叠百分比(默认值:0.7)
-f, --file FILE print results to file (default: stdout)##将结果打印到文件(默认:stdout)
--tab_table print tab-separated values table##打印制表符分隔的值表
-x, --extension EXTENSION
extension of bins (other files in directory are ignored) (default: fna)##Bin的扩展名(忽略目录中的其他文件)(默认:FNA)
-t, --threads THREADS
number of threads (default: 1)##线程数(默认为1)
--pplacer_threads PPLACER_THREADS
number of threads used by pplacer (memory usage increases linearly with additional threads) (default: 1)##Pplacer使用的线程数(内存使用量随着线程的增加而线性增加)(默认值:1)
-q, --quiet suppress console output##抑制控制台输出
--tmpdir TMPDIR specify an alternative directory for temporary files##为临时文件指定替代目录
Example: checkm lineage_wf ./bins ./outputcheckm的所有参数
usage: checkm
{data,tree,tree_qa,lineage_set,taxon_list,taxon_set,analyze,qa,lineage_wf,taxonomy_wf,gc_plot,coding_plot,tetra_plot,dist_plot,gc_bias_plot,nx_plot,len_hist,marker_plot,unbinned,coverage,tetra,profile,ssu_finder,merge,outliers,modify,unique,test}Overview一般不需要用到这块
Overview
Lineage-specific marker set##谱系特定的标记集
tree: place bins in the reference genome tree##把bins放到参考基因组树中
tree_qa: assess phylogenetic markers found in each bin##评估在每个bin中发现的系统发育标记
lineage_set: infer lineage-specific marker sets for each bin##为每个仓位推断特定于谱系的标记集
Taxonomic-specific marker set##特定于分类的标记集
taxon_list: list available taxonomic-specific marker sets##列出可用的特定于分类的标记集
taxon_set: infer taxonomic-specific marker set##推断特定于分类的标记集
Apply marker set to genome bins##将标记集应用于基因组bins
analyze: identify marker genes in bins##识别bins中的标记基因
qa: assess bins for contamination and completeness##评估bins的污染性和完整unbinned
usage: checkm unbinned [-h] [-x EXTENSION] [-s MIN_SEQ_LEN] [-q]
bin_dir seq_file output_seq_file output_stats_file
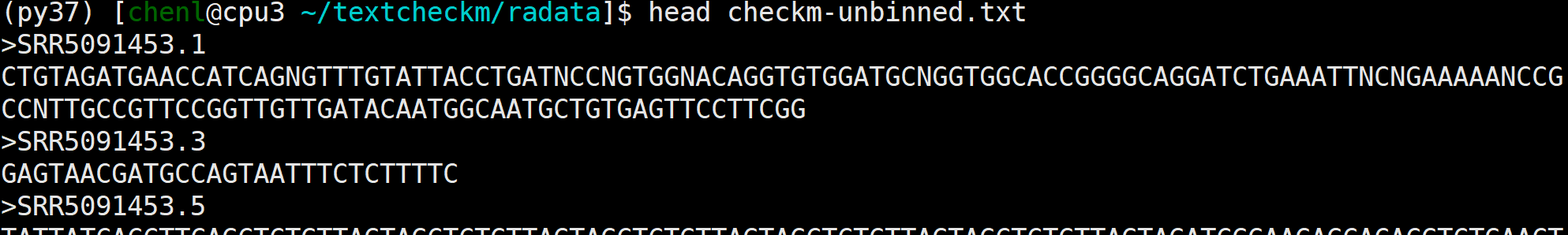
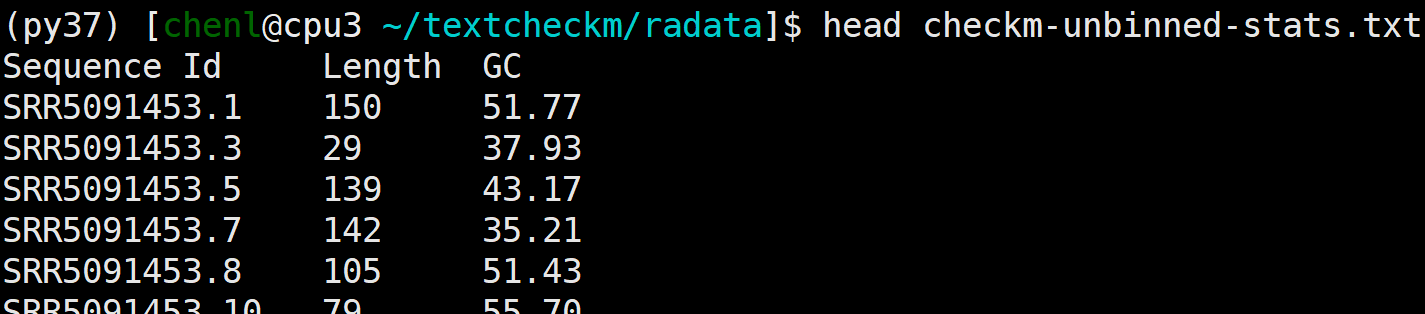
Example: checkm unbinned ./bins seqs.fna unbinned.fna unbinned_stats.tsv实战
(py37) [chenl@cpu3 ~/textcheckm/radata]$ checkm unbinned -x fa /home/chenl/textcheckm/SRR5091453/bin /home/chenl/textcheckm/radata/SRR5091453_1_paired.fasta checkm-unbinned.txt checkm-unbinned-stats.txt
##
coverage
usage: checkm coverage [-h] [-x EXTENSION] [-r] [-a MIN_ALIGN]
[-e MAX_EDIT_DIST] [-m MIN_QC] [-t THREADS] [-q]
bin_dir output_file bam_files [bam_files ...]
Example: checkm coverage ./bins coverage.tsv example_1.bam example_2.bamtetra
usage: checkm tetra [-h] [-t THREADS] [-q] seq_file output_file
Example: checkm tetra seqs.fna tetra.tsv实战
(py37) [chenl@cpu3 ~/textcheckm/radata]$ checkm tetra SRR5091453_1_paired.fasta SRR5091453-tetra.tsv
##结果
生成 SRR5091453-tetra.tsv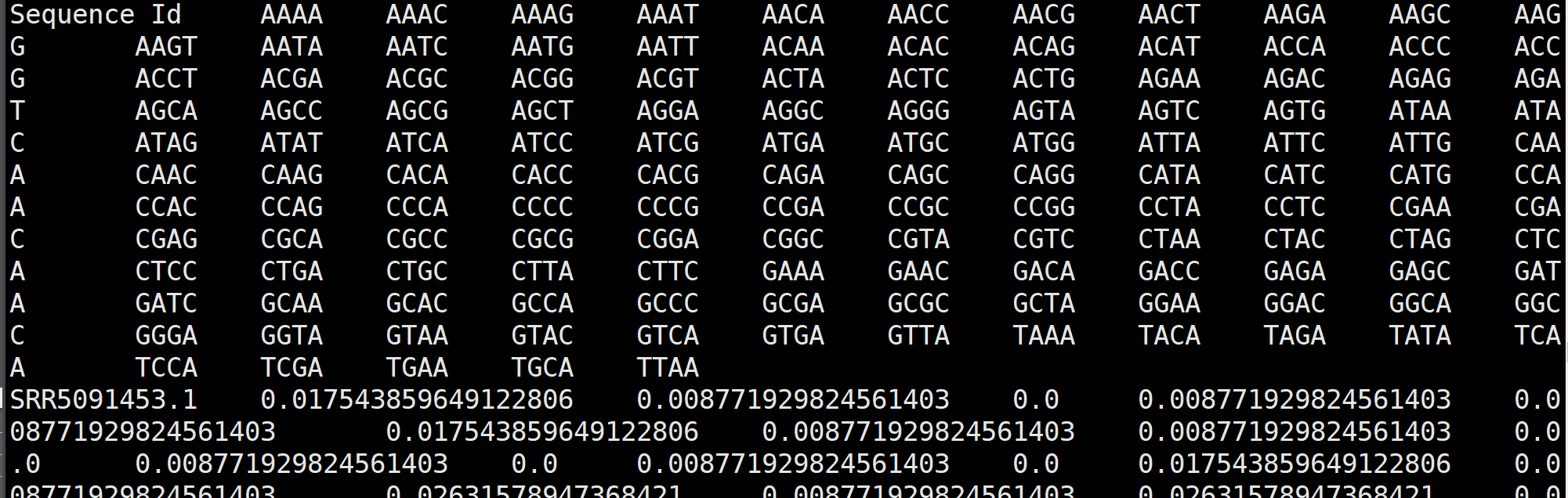
profile
usage: checkm profile [-h] [-f FILE] [--tab_table] [-q] coverage_file
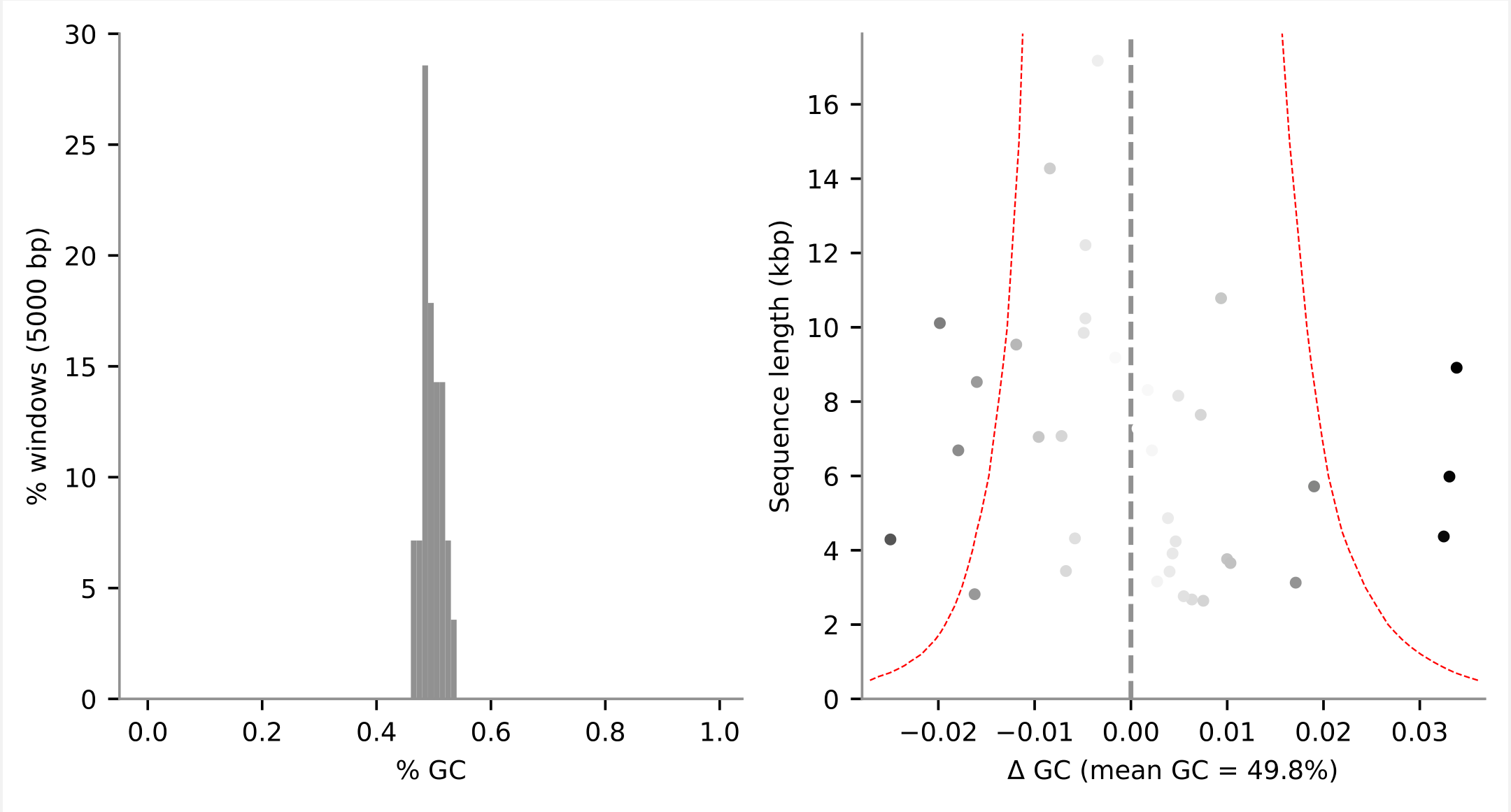
Example: checkm profile coverage.tsvgc_plot: create GC histogram and delta-GC plot
usage: checkm gc_plot [-h] [--image_type {eps,pdf,png,ps,svg}] [--dpi DPI]
[--font_size FONT_SIZE] [-x EXTENSION] [--width WIDTH]
[--height HEIGHT] [-w GC_WINDOW_SIZE] [-b GC_BIN_WIDTH]
[-q]
bin_dir output_dir dist_value [dist_value ...]##实战##有的参数没设置,dist_value随便设置的
checkm gc_plot --image_type pdf -x fa /home/chenl/textcheckm/SRR5091453/bin SRR5091453-gc_plot 50
##结果