🏃以下均为我个人在宏基因组学习过程中的笔记,参考性不大,参考链接很靠谱。🙋
有什么想找的请直接Ctrl + F (我写的很乱)😺
下面🏃这些链接是我在学习中的参考文章
https://www.jianshu.com/p/66ab14988a74
https://zhuanlan.zhihu.com/p/54080691
分箱Binning-MetaBAT2
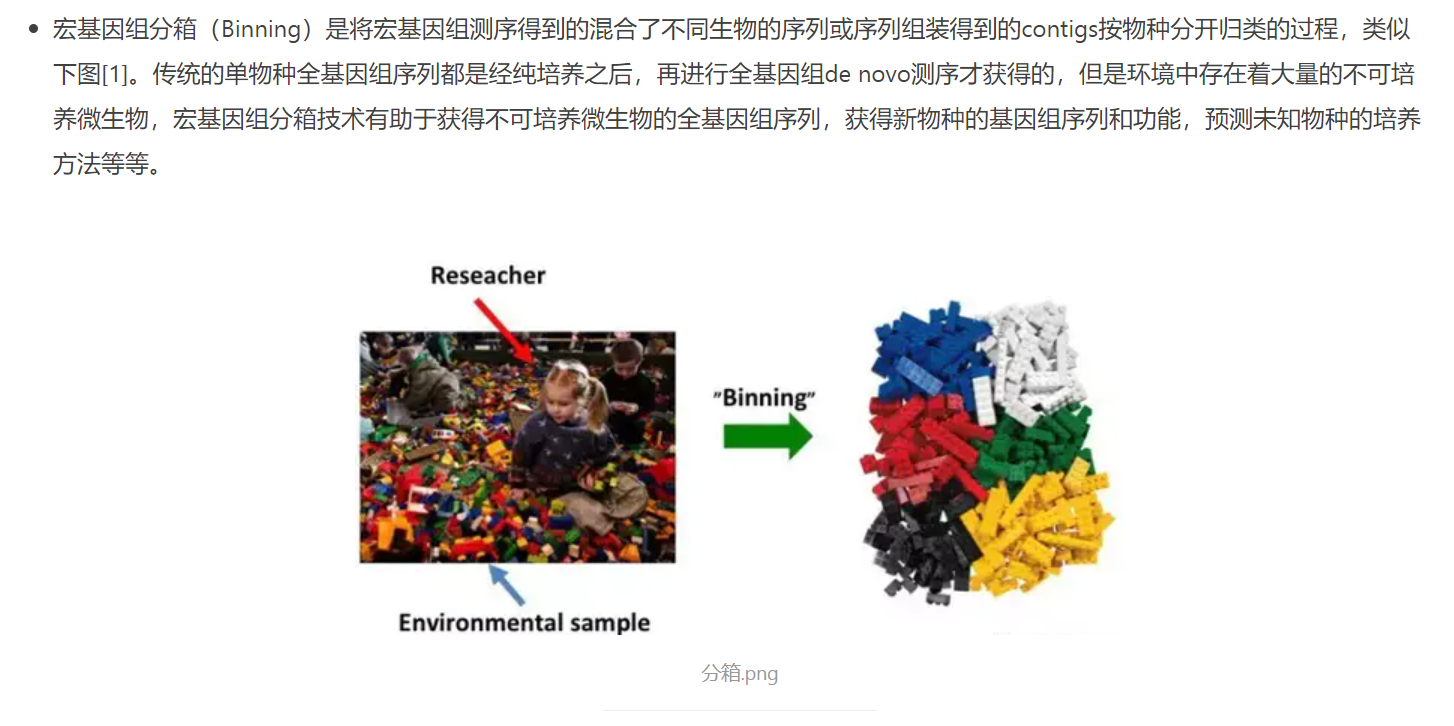
宏基因组分箱(Binning)是将宏基因组测序得到的混合了不同生物的序列或序列组装得到的contigs按==物种分开归类==的过程,类似下图。传统的单物种全基因组序列都是经纯培养之后,再进行全基因组de novo测序才获得的,但是环境中存在着大量的不可培养微生物,宏基因组分箱技术有助于获得不可培养微生物的全基因组序列,获得新物种的基因组==序列和功能==,预测未知物种的培养方法等等。
关联分析
即通过binning得到的bins(暂且简称为bins,更确切的说是strain-level clusters 或strain-level taxonomic units)可以进行宏基因组关联分析以及多组学联合分析,将特定功能代谢产物与特定物种、特定基因进行关联研究,推动其因果机制的探究,为疾病监控、环境监测提供了菌株水平的生物靶标
单菌组装
通过对binning得到的bins进行后续组装,可以得到很多不能在实验室里培养的细菌、古菌、病毒的基因组草图,然后根据单菌组装结果进行菌株水平的基因和功能注释、比较基因组分析、进化分析等,使我们得以洞察这些无法在实验室培养获得的菌株的生态适应机制,营养互作机制和新陈代谢功能等,可以研究在生态环境和复杂疾病中起重要作用的菌种以及致病菌和宿主的互作机制及其微进化机制。
工具选择:MetaBAT2(2022年3月)
MetaBAT2 被引用次数:602 MetaBAT2在单样本分箱中表现最佳
PeerJ 2019最好
MetaWRAP 被引用次数:413
MetaBAT 被引用次数:1151
PeerJCONCOCT 被引用次数:60 能获得最多的bin
MaxBin 2.0被引用次数:820 2017年Nature Methods的评估中最好
宏基因组最佳分箱工具Metabat2
实战
bowtie2-build --threads 30 /home/hanmz/project/RA/PRJNA356102/assembly/SRR5091489/SRR5091489.contigs.fa ./assembly/SRR5091489/SRR5091489-contigs_fabowtie2 -p 40 --local --very-sensitive-local -x ./assembly/SRR5091489/SRR5091489-contigs_fa -1 /home/hanmz/project/RA/PRJNA356102/rawdata/SRR5091489_1_paired.fastq.gz -2 /home/hanmz/project/RA/PRJNA356102/rawdata/SRR5091489_2_paired.fastq.gz -S ./assembly/SRR5091489/SRR5091489_contigs.samsamtools sort --threads 46 ./assembly/SRR5091489/SRR5091489_contigs.sam -o ./assembly/SRR5091489/SRR5091489_contigs.sort.bamjgi_summarize_bam_contig_depths --outputDepth ./assembly/SRR5091489/SRR5091489_depth.txt ./assembly/SRR5091489/SRR5091489_contigs.sort.bammetabat2 -i /home/hanmz/project/RA/PRJNA356102/assembly/SRR5091489/SRR5091489.contigs.fa -a ./assembly/SRR5091489/SRR5091489_depth.txt -o ./assembly/SRR5091489/bin/SRR5091489_bin -t 46 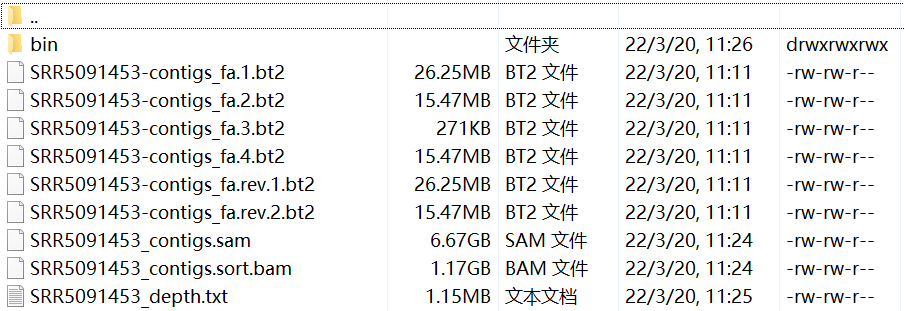
得到以上文件
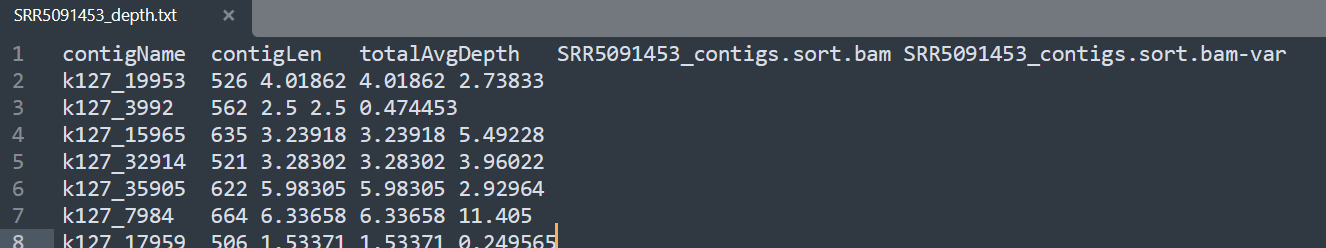
_depth.txt文件为统计contig深度得到的文件
bin目录下为.fa文件,==为分箱结果==
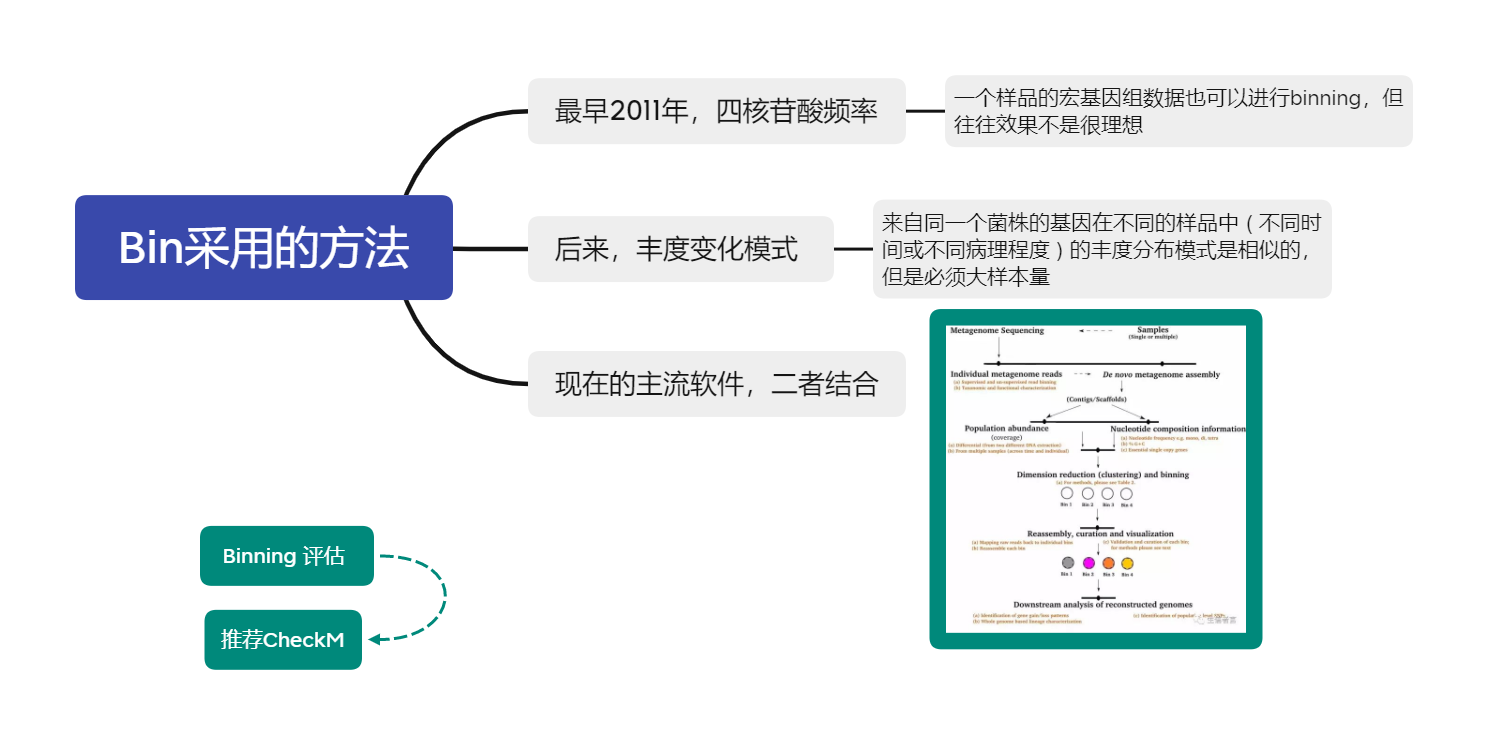
Binning评估-CheckM
该软件的评估结果会给出==completeness==和==contamination==两个指标。同时也可以结合常用来检验样品污染的GC-Depth分布图来看是否存在污染,是否是chimeric genome bins。
相关链接
checkM的GitHubhttps://github.com/Ecogenomics/CheckM
checkM的databasehttps://data.ace.uq.edu.au/public/CheckM_databases/
checkM说明文档https://github.com/Ecogenomics/CheckM/wiki
安装指导:
https://shenmengyuan.github.io/2016/12/22/checkm/相当详细😍⭐:raising_hand:
CheckM官方文档有道翻译
CheckM提供了一套工具,用于==评估==从分离物、单细胞或宏基因组中恢复的基因组的==质量==。
它通过使用在系统进化谱系中普遍存在的==单拷贝的基因组合==,提供了对基因组==完整性和污染的可靠估计==。
基因组质量的评估也可以使用描绘关键基因组特征(例如,GC,编码密度)的图来检查,这些图突出了典型基因组预期分布之外的序列。
CheckM还提供了一些工具,用于根据标记集的兼容性、基因组特征的相似性和参考基因组树的邻近性来识别可能需要合并的基因组箱。
如果你计划处理大量的基因组,你可能希望把它们分成小批量。
在64GB的机器上,用40个线程同时运行1000个基因组,运行得很好。
超过机器的可用内存将导致checkkm使用交换空间(与任何程序一样),这将大大增加处理基因组的时间。
Bioinformatic tool dependencies
CheckM requires the following programs to be added to your system path:
HMMER (>=3.1b1)
安装之后还有几步需要操作,可以看下载的帮助文档
prodigal (2.60 or >=2.6.1)
pplacer (>=1.1)guppy是pplacer的一部分也必须添加到环境变量中
推荐使用pip安装CheckM:
pip3是在Python3中的pip
pip3 install numpy
pip3 install matplotlib
pip3 install pysam
pip3 install checkm-genomeRequired reference data参考数据库的下载
CheckM relies on a number of precalculated data files which can be downloaded from https://data.ace.uq.edu.au/public/CheckM_databases/. Decompress the file to an appropriate folder and run the following to inform CheckM of where the files have been placed:
> checkm data setRoot <checkm_data_dir>#测试数据
~/biosoft/metabat_data
$ checkm test ~/checkm_test_results
####结果
[2022-03-31 19:57:20] INFO: [Passed]
[2022-03-31 19:57:20] INFO: { Current stage: 0:00:00.005 || Total: 0:05:17.882 }
1. 简介
基因组组装或者宏基因组分箱 (binning) 获得的草图数据,首先需要评估其质量,包括基因组完整度、污染度、序列分布等信息。最用的软件有 CheckM。
CheckM 提供了一系列工具用于评估从分离培养、单细胞、宏基因组获得的基因组质量,可以根据基因组在参考基因组发育树中的位置来推断其精确的单拷贝标记基因集 (lineage-specific marker set),同时也提供数据库可用的基于分类学的基因集 (taxonomic-specific marker set)。
CheckM 利用基因的单拷贝性来有效的估计基因组完整度和污染可能性,同时能绘制基因组关键特征 (例如:GC含量) 的图像来评估基因组的质量。
还可以进一步从每一个分箱中找出16S rRNA基因,以便于后续对所拼装达到的菌种的分类信息进行鉴定。
(py37) [chenl@cpu4 ~]$ whereis checkm
checkm: /home/chenl/miniconda2/envs/py37/bin/checkm实战
checkm lineage_wf -t 45 -x fa --nt --tab_table -f /home/chenl/RA/PRJNA356102/assembly/SRR5091489/result-bin/SRR5091489_bins_qa.txt /home/chenl/RA/PRJNA356102/assembly/SRR5091489/bin /home/chenl/RA/PRJNA356102/assembly/SRR5091489/result-bin求求给个三连再走
📢🙇🙏:+1:⭐💓
checkm lineage_wf -t 46 -x fa --nt --tab_table -f bins_qa.txt metabat_bins bin_qa_result 完整度是实际上有的Mark基因除以应该有的基因
污染度是bin里包含的基因有多少不属于这个bin的(幅度大)
checkm unbinned
