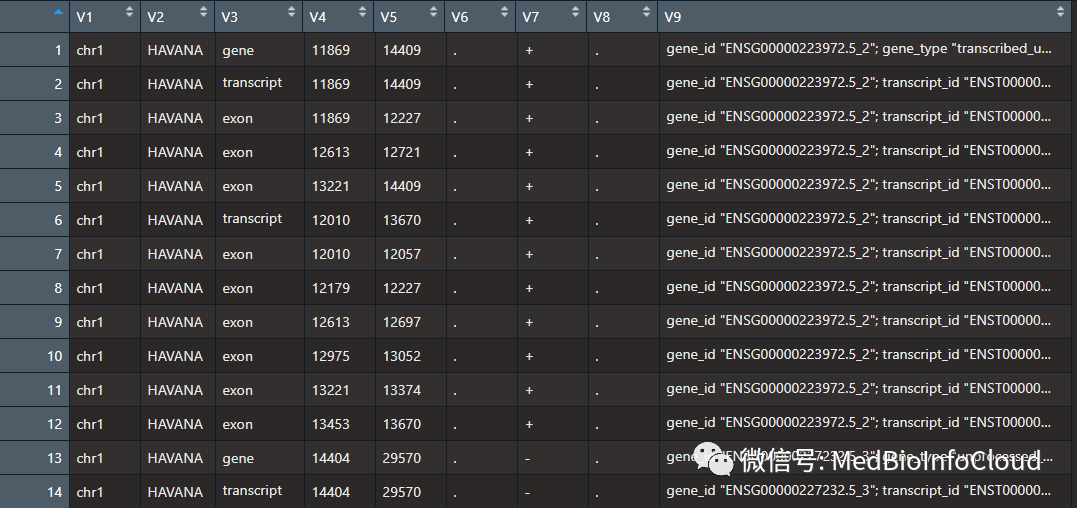
Common Data File Formats in Bioinformatics
利用Sanger、illumina、第三代和第四代测序技术,我们测序得到的是带有质量值的碱基序列==fastq==格式,参考基因组是==fasta==格式。
用比对工具把fastq格式的序列回帖到对应的fasta格式的参考基因组序列,就可以产⽣==sam==格式的⽐对⽂件。
把sam格式的⽂本⽂件压缩成==⼆进制bam==⽂件可以节省空间。
如果是记录某些位点或者区域碱基的变化,就是==VCF==⽂件格式。
如果对参考基因组上面的各个区段标记它们的性质,比如哪些区域是外显子,内含子, UTR等等,这就是==gtf/gff==格式。
如果只是为了单纯描述某个基因组区域,就是==bed==格式⽂件,记录染色体号以及起始终止坐标,正负链即可。
fastq文件
概念
fastq=fasta+quality(Fasta格式加上质量数据)
https://support.illumina.com/bulletins/2016/04/chinese-fastq-files-explained.html
https://help.basespace.illumina.com/articles/descriptive/fastq-files/
Illumina测序技术使用 簇生成和边合成边测序(SBS)化学技术对流动槽(flow cell)上数百万或数十亿簇(cluster)进行测序,具体簇的数目取决于测序平台。 在边合成边测序化学过程中,仪器上的实时分析(RTA)软件对每个簇的每个循环进行碱基检出和存储。 RTA以单个读取碱基(base call,或称BCL)文件的形式存储碱基检出数据。 测序完成后,必须将BCL文件中的测定的碱基转换为序列数据。 此过程称为BCL到FASTQ的转换。
FASTQ是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。其序列以及质量信息都是使用一个ASCII字符标示,最初由Sanger开发,目的是将FASTA序列与质量数据放到一起,目前已经成为高通量测序结果的事实标准。
对于单端测序的运行,将为每个流动槽上每条通道的每个样品创建一个Read 1(R1)FASTQ文件。 对于双端测序的运行,将为每个流动槽上每条通道的每个样品各创建一个R1和一个Read 2(R2)FASTQ文件。 FASTQ文件是使用扩展名*.fastq.gz压缩和创建的。
格式


第一行以@开头,序列标识符,其中包含有关测序运行和簇的信息。 该行的具体内容会因使用的BCL到FASTQ转换软件而不同。(与FASTA格式的描述行类似)
序列(碱基信号; A,C,T,G和N)。
分隔符,只是一个加号(+),之后可以再次加上序列的标识及描述信息(可选)。
读取碱基的质量值。,与第二行的序列相对应,长度必须与第二行相同。这些是Phred +33编码的,使用ASCII字符表示数字质量值。
其中!为最低质量、~则为最高质量。以下字符从左到右代表从低到高的质量得分的:
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~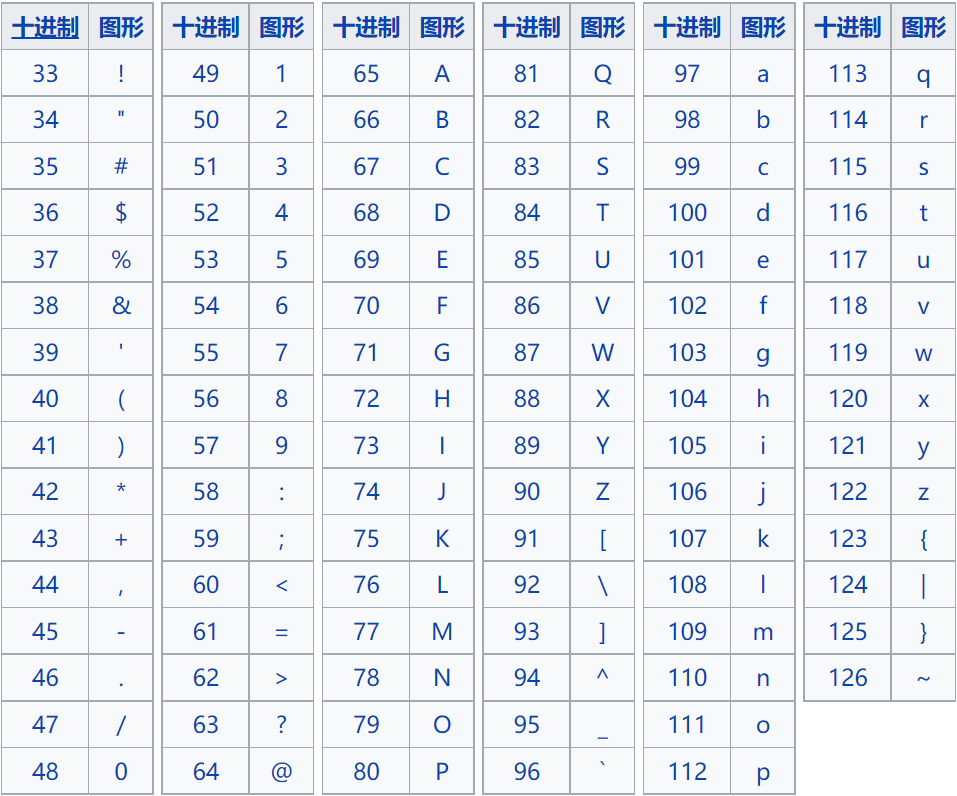
质量值的计算公式如下:$Q=-10 \times log_{10}P$
这个公式中Q表示的是潜在错误的概率,Q值越大,表明测错的可能性越小。下面是Q值对应的错误率表:
| Phred Quality Score | Probability of incorrect base call | Base call accuracy |
|---|---|---|
| 10 | 1 in 10 | 90% |
| 20 | 1 in 100 | 99% |
| 30 | 1 in 1000 | 99.9% |
| 40 | 1 in 10000 | 99.99% |
| 50 | 1 in 100000 | 99.999% |
| 60 | 1 in 1000000 | 99.9999% |
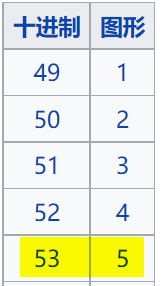
举个犊子,假设==质量信息是5==,这个5看成字符,不要看成数字,那么他对应的十进制数就是53,如果使用的是Sanger测序,那么质量评分采用的就是Phred+33的形式,那么Q = 53-33=20=-10lg(errorP),所以errorP = 0.01。也就计算出错误率啦,就便于我们进行质控。每一个碱基都有一个质量评分,所以第2行和第4行的位数是相同的。==Q值=20>>errorP=0.01>>碱基准确度就是99%。==
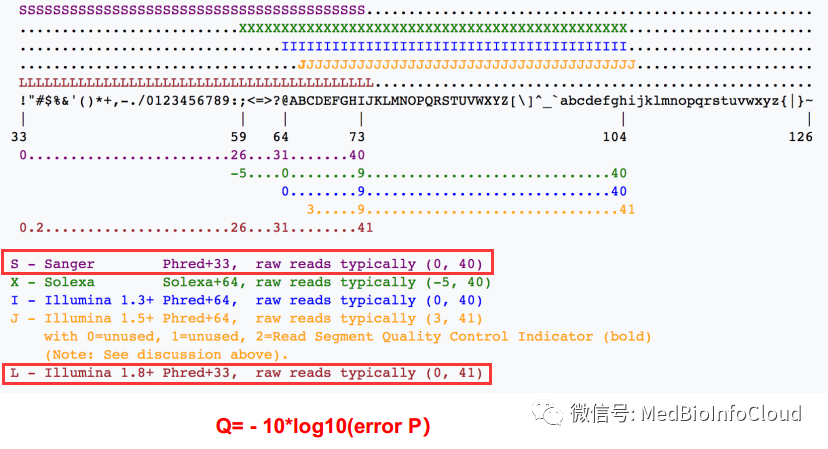
质量评分指的是一个碱基的错误概率的对数值。其最初在Phred拼接软件中定义与使用,对于每个碱基的质量编码标示,不同的软件采用不同的方案,目前有5种方案:
- Sanger,Phred quality score,值的范围从0到92,对应的ASCII码从33到126,但是对于测序数据(raw read data)质量得分通常小于60,序列拼接或者mapping可能用到更大的分数。
- Solexa/Illumina 1.0, Solexa/Illumina quality score,值的范围从-5到63,对应的ASCII码从59到126,对于测序数据,得分一般在-5到40之间;
- Illumina 1.3+,Phred quality score,值的范围从0到62对应的ASCII码从64到126,低于测序数据,得分在0到40之间;
- Illumina 1.5+,Phred quality score,但是0到2作为另外的标示;
- Illumina 1.8+ ,Phred quality score。
fasta文件
概念
FASTA格式是一种用于表示==核苷酸序列==或==多肽序列==的文本格式。其中碱基对或氨基酸用==单个字母==来表示,且允许在序列前添加序列名及注释。该格式已成为生物信息学领域的一项标准。
FASTA简明的格式降低了序列操纵和分析的难度,令序列可被文本处理工具和诸如Python、Ruby和Perl等脚本语言处理。
格式
FASTA格式中的一条完整序列,包含开头的单行描述行和多行序列数据。描述行行首前置半角大于号(“>”)以和数据行区分。“>”后紧接的内容为该序列的标识符(==唯一的==),该行剩余部分则为序列的描述(标识符与描述均非必须)。“>”和标识符之间不应有空格,且建议将单行内容限制在80字符以内。序列的结束以下一条序列的“>”出现为标识。如下为FASTA评论区格式一条序列的示例:
>gi|31563518|ref|NP_852610.1 关,瓦斯4 microtubule-associated proteins 1A/1B light chain 3A isoform b [Homo sapiens]
MKMRFFSSPCGKAAVDPADRCKEVQQIRDQHPSKIPVIIERYKGEKQLPVLDKTKFLVPDHVNMSELVKI
IRRRLQLNPTQAFFLLVNQHSMVSVSTPIADIYEQEKDEDGFLYMVYASQETFGFIRENE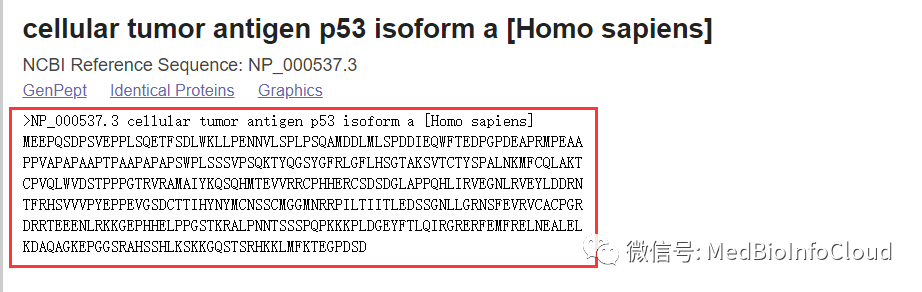
核苷酸序列:

氨基酸序列:

SAM文件/BAM文件
概念
当我们测序得到的fastq数据map到基因组(比如用bowtie2来mapping)之后,会得到一个以sam或bam为扩展名的文件。这里,SAM的全称是sequence alignment/map format。而BAM就是SAM的二进制文件,也就是压缩格式的sam文件。
SAM格式文件包括头部注释部分和比对结果部分,头部分为’’可选部分’’。头部分位于比对部分之前,以“@”开头。比对部分有11列是固定的,其他多列可选。
头部信息介绍
每个标题行以字符”@”开头,后面是两个字母的记录类型代码。在标题中,每一行都是由制表符分隔的。除了@CO行,每个数据字段都遵循格式”TAG:VALUE”,其中TAG是两个字母组成的字符串,定义了内容和值的格式。每个标题行应该匹配:/^ @(HD|SQ|RG|PG)(t[A-Z/a-z][A-Z/a-z/0-9]:[-~]+)+$ /或/^@CO\t.*/。小写字母是保留给用户自定义内容使用的,不会在任何版本定义中出现。以下给出了定义的记录类型和标记。当以下TAG标签出现”*“时,表示该TAG是必须的,如@SQ必须有SN和LN字段。
比对信息介绍
比对信息部分(alignment section),每一行表示一个片段(segment)的比对信息,包括11个必须的字段(mandatory fields)和一个可选的字段,字段之间用tab分割。必须的字段有11个,顺序固定,不可用时,根据字段定义,可以为’0’或者’*’,这是11个字段包括:
1. QNAME 比对片段的(template)的编号;read name,read的名字通常包括测序平台等信息
# eg.E00528:375:HN2JTCCXY:8:1123:5457:24479
2. FLAG 位标识,template mapping情况的数字表示,每一个数字代表一种比对情况,这里的值是符合情况的数字相加总和
# eg.16
3. RNAME 参考序列的编号,如果头部注释中对SQ-SN进行了定义,这里必须和其保持一致,另外对于没有mapping上的序列,这里是'*';
# eg.chr1
4. POS 比对上的位置,注意是从1开始计数,没有比对上,此处为0;
# eg.36576599
5. MAPQ mapping的质量,比对的质量分数,越高说明该read比对到参考基因组上的位置越唯一;
# eg.42
6. CIGAR 简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),其以参考序列为基础,使用数字加字母表示比对结果,match/mismatch、insertion、deletion 对应字母 M、I、D。比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
# eg.36M 表示36个碱基在比对时完全匹配
###注:第七列到第九列是mate的信息,若是单末端测序这几列均无意义###
7. RNEXT 配对片段(即mate)比对上的参考序列的编号,没有另外的片段,这里是'*',同一个片段,用'=';
# eg.*
8. PNEXT 配对片段(即mate)比对到参考序列上的第一个碱基位置,若无mate,则为0;
# eg.0
9. TLEN Template(文库插入序列)的长度,最左边的为正,最右边的为负,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,此处为0(ISIZE,Inferred fragment size.详见Illumina中paired end sequencing 和 mate pair sequencing,是负数,推测应该是两条read之间的间隔(待查证),若无mate则为0);
# eg.0
10. SEQ 序列片段的序列信息,如果不存储此类信息,此处为'*',注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度;
# eg.CGTTTCTGTGGGTGATGGGCCTGAGGGGCGTTCTCN
11. QUAL 序列的质量信息, read质量的ASCII编码。格式同FASTQ一样。
# eg.F-?DD<,EEFFEEC?F5FA7FAF8FAF;F@E:DF>FEEAFFFFFCFFC4EFDFDDFEFE=EFFF
12.第十二列及之后:Optional fields,以tab建分割。
# eg.AS:i:-1 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:35T0 YT:Z:UU\1. **QNAME**:reads名称。
\2. **FLAG**:reads比对情况。不同的情况对应不同的值,这里的数字是所有情况的和。
\3. **RNAME**:比对至参考序列的名称。
\4. **POS**:比对到的位置。
\5. **MAPQ**:比对质量。
\6. **CIGAR**:比对情况信息。
\7. **RNEXT**:与之配对的另一条reads所在的参考序列名称。"="表示位于同一个参考序列上,"*"表示没有另一条reads。
\8. **PNEXT**:与之配对的另一条reads所在的位置。
\9. **TLEN**:插入片段长度。
\10. **SEQ**:reads序列。
\11. **QUAL**:reads序列质量
NH:i:n 表示reads比对到参考序列位置的个数。
AS:i:n 表示比对得分。格式
例
@HD VN:1.4 SO:coordinate@HD VN:1.5 SO:unsorted GO:query ##输出文件第一行
@SQ SN:k127_19953 LN:526
@SQ SN:k127_3992 LN:562 ###@SQ 参考序列字典,@SQ行的顺序定义了比对文件排序顺序。
@SQ SN:k127_15965 LN:635 ###SN* 参考序列名字(染色体)。用于RNAME和PNEXT比对的记录
@SQ SN:k127_32914 LN:521 ###LN* 参考序列长度,范围:[$1,2^{31}-1$]
@SQ SN:k127_35905 LN:622 ###
@SQ SN:k127_7984 LN:664 ###
@SQ SN:k127_17959 LN:506 ###
@SQ SN:k127_20950 LN:547 ###
@SQ SN:k127_11976 LN:697 ###
@SQ SN:k127_26932 LN:770 ###
@SQ SN:k127_2995 LN:515 ###
@SQ SN:k127_23941 LN:826 ###
@SQ SN:k127_41887 LN:788 ###
@SQ SN:k127_14968 LN:970 ###
@SQ SN:k127_8983 LN:511 #### 例如
@SQ SN:chr1 LN:249250621 M5:1b22b98cdeb4a9304cb5d48026a85128 UR:file:/opt/biosoft/ref/sv/hg19_ref_genome.primary-assembly.faE00514:173:H3C3JCCXY:4:1124:12398:67234 337 Chr00 32904 0 150M Chr09 33498107 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:0 RG:Z:J36CK1
E00514:173:H3C3JCCXY:4:1124:12398:67234 369 Chr00 32904 0 150M Chr16 2469225 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:2 RG:Z:J36CK1
E00514:173:H3C3JCCXY:4:1124:12398:67234 369 Chr00 32904 0 150M Chr16 29515410 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:4 RG:Z:J36CK1GTF和GFF文件
概念
GTF全称是Gene transfer format,用于储存基因结构信息,总共有9列。关于gtf文件,我在文章:生信中各种ID转换中就已经有所应用。我之前在TCGA数据库差异分析的文章中,也是通过gtf文件进行ID转换的。
格式